细胞可塑性是指细胞对内在或外在因素的反应,转而重编程并改变其命运和身份的能力。可塑性不仅仅是干细胞的特征,细胞可以通过去分化(在同一谱系中将分化的细胞逆转为未分化状态)、转分化(将分化的细胞转化为另一分化的细胞谱系)以及上皮-间充质转化(EMT)并获得不同的表型。
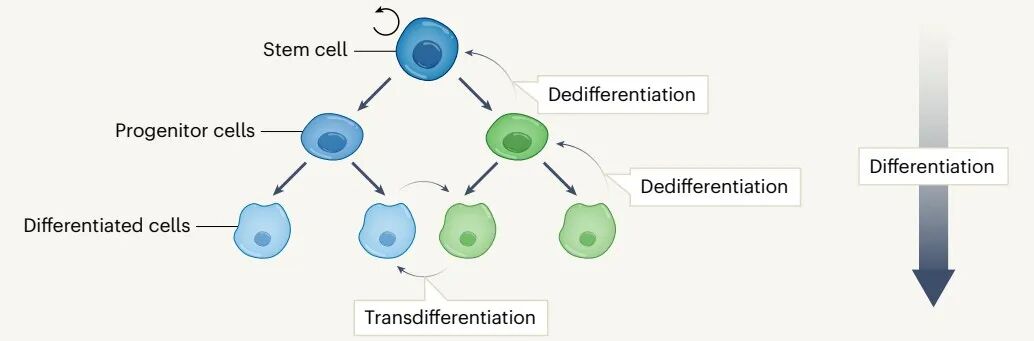
可塑性对组织损伤、炎症或衰老后恢复体内平衡至关重要,但也可能导致肿瘤发生。在癌症进展过程中,肿瘤细胞可以在细胞状态之间切换,这一过程主要由细胞可塑性介导,以克服选择性压力。因此,细胞可塑性在很大程度上促进了肿瘤内的异质性和适应性,并对肿瘤生长、转移和治疗耐药性产生重大影响。因此,了解驱动细胞可塑性的内在和外在机制,以及这些机制如何促进肿瘤生长、增殖、转移和药物耐受,对我们如何利用细胞可塑性进行抗癌治疗至关重要。
分化细胞恢复到干细胞样状态的能力对肿瘤发生具有重要意义,一些致癌驱动因素影响肿瘤发生过程中的可塑性。抑癌基因如TP53、RB1和PTEN调节发育分化程序,当它们功能失调时,导致癌症发生。
例如在腺上皮细胞中,单能基底干细胞和管腔干细胞可以在肿瘤发生过程中重新获得多能性。在小鼠前列腺肿瘤发生过程中,基底细胞中的Pten缺失促进基底向管腔进而向肿瘤细胞的转分化。在小鼠乳腺癌中,磷脂酰肌醇-4,5-二磷酸3-激酶催化亚基α(Pik3ca)H1047R的表达在肿瘤发生早期诱导祖细胞的多能性,为肿瘤内异质性奠定了基础。
炎症也调节再生和肿瘤发生过程中的可塑性。在小鼠小肠中,炎症后Lgr5+干细胞丢失,从而诱导Paneth细胞重新进入细胞周期,获得干细胞样特性,并有助于组织再生。在没有炎症的情况下,只有肠道干细胞才能在Apc缺失后诱导肿瘤形成。而Apc和Nfkbia的共同缺失激活了NF-κB信号传导,诱导非干细胞形成肿瘤,表明炎症信号可以扩大其肿瘤起始能力。
癌症干细胞(CSCs)表达干细胞样程序,能够自我更新,维持肿瘤生长,并产生增殖能力更受限制的肿瘤细胞。在严格的意义上讲,CSC会产生生长和分化潜力更有限的亚群,并永远不会恢复到CSC状态。然而,有证据表明,CSC和非CSC都是可塑性的,并且可能在某些条件下发生表型转变。例如,异种移植小鼠癌症类器官中Lgr5+CSCs的基因敲除限制了肿瘤生长而不会导致消退。然后,肿瘤由补充CSC池的增殖性Lgr5-细胞维持。当敲除停止时,Lgr5+CSC再次出现,导致肿瘤快速再生,并表明CSC敲除后分化程度更高的肿瘤细胞具有可塑性。这一发现得到了患者来源类器官的支持,这表明靶向CSCs而不防止细胞可塑性的治疗是不够的。
CSC生态位由异质性和相互作用的细胞群组成,在肿瘤发生中起主要作用,对CSC调节和促进癌症细胞可塑性至关重要。血管生态位是指由内皮细胞、周细胞、平滑肌细胞和免疫细胞组成的一个专门的高度血管化区域,它通过影响干性、化疗耐药性、侵袭和转移来创造一个允许肿瘤的微环境。
内皮细胞通过分泌Wnt和Notch配体以及直接的细胞-细胞相互作用来维持CSC中的干性,内皮细胞还通过IL-8和IL-6的分泌增加侵袭性和增殖。而CSCs可以通过分泌血管内皮生长因子(VEGF)诱导血管小丛形成,进而调节CSC的更新。
除了在肿瘤发生过程中吸引和重编程内皮细胞外,CSCs还可以通过模拟血管转分化为内皮样细胞。肿瘤内的低氧水平可能促进CSCs干性并获得内皮特征,肿瘤细胞向内皮细胞的分化已在不同的人类和小鼠癌症模型中得到证实,但其生物学相关性尚不清楚。CAFs通过细胞因子分泌参与CSC维持,包括HGF、IGF2、TGFβ1、IL-6和多种趋化因子配体,并通过基质金属蛋白酶分泌和胶原和透明质酸沉积参与基质重塑。
免疫细胞也是CSC生态位的关键组成部分。CSC和巨噬细胞的通讯通过直接相互作用发生,其中巨噬细胞产生的CSC生态位促进了EMT,诱导CSC中的EphA4表达,这反过来促进细胞因子分泌并维持CSC干性。巨噬细胞分泌的细胞因子(例如TGF-β、IL-6、Wnt配体)主要通过STAT-3信号传导促进肿瘤细胞的干性。
转移是通过多步级联发生的,包括癌症细胞从原发肿瘤脱离、局部侵入周围组织、侵入血管或淋巴管、外渗、继发器官定植和继发肿瘤生长。越来越多的证据表明,只有某些肿瘤细胞亚群,称为转移起始细胞(MIC),能够形成转移。MIC具有高度可塑性,在整个转移级联过程中表现出不同程度的干性、EMT和代谢可塑性。

转移起始
EMT对肿瘤转移非常重要,EMT可以由不同的转录因子触发,SNAI1、SNAI2、Twist1、ZEB1和ZEB2被认为是EMT的核心转录因子,可以诱导经典的EMT程序,并且通常是共表达的。EMT促进干性,使MIC产生继发性肿瘤。
在肿瘤发生过程中,癌症细胞的代谢表型可以根据营养物质的可利用性、增殖率和肿瘤突变负担而改变。转移级联增加了转移肿瘤细胞的适应性,以克服营养变化和氧化应激,MIC通常表现为厌氧糖酵解增加。在多种癌症中发生氧化磷酸化失调,并与EMT和预后不良相关。
生态位对于EMT诱导和转移起始至关重要。成纤维细胞通过分泌细胞外基质和基质金属蛋白酶来支持肿瘤细胞,促进迁移、侵袭和血管生成,并有利于肿瘤细胞的可塑性。肿瘤细胞分泌TGF-β对成纤维细胞在肿瘤发生中第一步的募集和激活至关重要。活化的成纤维细胞随后激活TGF-β的自分泌和旁分泌,在肿瘤细胞中诱导EMT并促进免疫逃逸。此外,巨噬细胞也影响EMT和肿瘤细胞的可塑性。
肿瘤细胞的局部侵袭和播散
处于完全EMT状态的肿瘤细胞侵入其周围组织,而混合EMT状态促进集体迁移,处于前沿的肿瘤细胞表现出比跟随细胞更明显的EMT表型。混合EMT细胞集体迁移与可塑性、干性、侵袭性和转移能力增加有关。接下来,肿瘤细胞作为循环肿瘤细胞(CTC)侵入血管,其中一些细胞存活下来,外渗到第二器官。在第二器官中,它们将增殖以实现转移性生长或经历休眠。
单个和聚集性CTC都表现出上皮和间充质标志物表达的变化,在肿瘤进展过程中表现出可塑性。不同CTC表型的可塑性已被证明有助于癌症的进展和化疗耐药性。在循环中,CTC的氧化应激增加,为了防止活性氧(ROS)介导的细胞死亡,肿瘤细胞增加抗氧化剂的产生。在黑色素瘤中,通过血管迁移的CTC比淋巴管中的CTC受到更高的氧化应激和铁死亡,并依赖铁死亡抑制剂GPX4生存,而通过淋巴管迁移的CTCs依赖抗氧化剂,如油酸和谷胱甘肽。
肿瘤细胞通过被血小板包裹并与白细胞、成纤维细胞、巨噬细胞和内皮细胞相互作用而在血液中存活。肿瘤细胞和巨噬细胞之间的交叉作用是CTC介导的结直肠癌转移所必需的,并促进EMT相关的可塑性。中性粒细胞-肿瘤细胞簇似乎比单独的肿瘤细胞簇更具转移性,这是由于肿瘤细胞中中性粒细胞介导的细胞周期进展增加。与血小板的相互作用提供了对血流粉碎力的抵抗力,并通过TGF-β和NF-κB途径激活诱导EMT。
转移生态位
转移生态位是由基质细胞、细胞外基质和刺激转移形成的扩散信号产生的特定微环境。越来越多的证据表明,肿瘤细胞在定植之前就准备好了它们的生态位。转移前生态位调节涉及血管渗漏、驻留细胞的重编程和骨髓衍生细胞的募集。一些机制是由转移部位的播散细胞诱导的,但原发性肿瘤也会通过分泌可溶性分子和外泌体进行远距离重编程。重编程的血管周围细胞表现出细胞外基质成分的增殖和表达增加,为转移创造了一个允许的土壤。
转移性定植
间充质-上皮转化(MET)的EMT逆转可以促进转移。E-钙粘蛋白的缺失增加了侵袭性,但其表达保护细胞在传播过程中免受氧化应激,促进转移定植。肿瘤细胞可以使用转移生态位中基质细胞表达的E-钙粘蛋白和N-钙粘蛋白形成异型连接,促进生存和生长。
几项研究强调了下调EMT因子对转移形成的必要性。鳞状细胞癌中Twist1介导的EMT促进侵袭和CTC循环,而Twist1下调促进转移定植。PRRX1促进EMT和胰腺导管腺癌的侵袭,PRRX1的作用后来被证明是由两种不同的亚型介导的:PRRX1b促进EMT、侵袭和迁移,PRRX1a刺激肝转移生长、肿瘤分化和MET。因此,转移性传播需要从转移级联的第一步的PRRX1b切换到后期的PRRX1a。
肿瘤休眠
播散的肿瘤细胞可以在转移部位进入休眠状态。这种生长停滞是由于血管形成不良、免疫破坏、缺乏营养和生长因子或通过来自微环境的抑制信号(例如TGF-β)而导致的增殖和凋亡之间的平衡。休眠细胞的特征是激活的生存途径、细胞周期停滞、持续的未折叠蛋白反应和低氧。休眠使细胞能够逃避免疫反应和化疗,通过成像技术仍然无法检测到,但即使在临床缓解几年后,也会导致复发。
肿瘤细胞进入和退出休眠的机制尚不完全清楚。休眠细胞在状态之间转换时表现出可塑性,但EMT或MET是否能促进再激活和从休眠中唤醒仍不清楚。休眠受到微环境的严格控制。转移部位的肿瘤细胞分泌III型胶原有利于休眠,而富含III型胶原的基质的破坏通过盘状蛋白结构域受体酪氨酸激酶1介导的STAT1信号传导诱导休眠细胞的觉醒和增殖。衰老过程中微环境的改变也在进入或退出休眠中发挥作用,成纤维细胞的年龄相关变化与黑色素瘤转移增加有关。衰老的成纤维细胞显示出Wnt拮抗剂sFRP2的分泌增加,其在黑色素瘤细胞中诱导对ROS介导的DNA损伤反应的抵抗,赋予对治疗的抵抗力并增加转移。肺中的老年成纤维细胞分泌更多的sFRP1并阻断Wnt5a介导的休眠诱导,刺激转移生长。影响微环境的年龄相关变化可能解释了治疗多年后转移性病变的复发。
肿瘤细胞可塑性在癌症发生和发展、转移和治疗耐药性中发生着关键的作用。不同的可塑性模式通过增殖状态和CSCs参与维持肿瘤生长,这在转移级联中也是必不可少的。可塑性还允许肿瘤细胞逃避选择性压力并耐药。因此,更好地了解肿瘤细胞调节可塑性的内在和外在机制,可以为新的治疗策略开辟道路,并在不久的将来提高患者的生存率。
参考文献:
1.Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat Cancer.2023 Aug 3



